Analyzing the simulation results
 First we are going to start the pulling-simulations on streptavidin-biotin, so they have time to finish while you learn how to set up an MD simulation from scratch in part 1 of the exercise.
First we are going to start the pulling-simulations on streptavidin-biotin, so they have time to finish while you learn how to set up an MD simulation from scratch in part 1 of the exercise.
To start the simulations, ssh to the daint-cluster:
ela ssh daint
Once done, load the daint hybrid modules and gromacs, the simulation program that we are going to use today.
module load daint-gpu module load GROMACS
To make copying the simulation results to your local machine easier, create a new folder in which all simulations will be carried out.
mkdir gromacs_tutorial cd gromacs_tutorial
The simulation input-files have already been prepared for you. You can download them using this little script. Download the script, e.g. using wget
wget http://www2.mpibpc.mpg.de/groups/grubmueller/Lugano_Tutorial/part2/download_files.bash
Then make the script executable and use it to download the other files.
chmod +x download_files.bash bash download_files.bash
You will find that a few files were downloaded and two directories, "pull1" and "pull2", were created. Each folder contains a set of MD parameters (.mdp-file) and a jobscript that we are going to use later to run the simulation on the cluster. Before we can do that, we need to run the gromacs-preprocessor for each simulation.
cd pull1 gmx grompp -f pull1.mdp -c ../start.gro -p ../topol.top -n ../index.ndx -o pull1.tpr
Change the names to "pull2" in the last line and also run the preprocessor (gmx grompp) in "pull2".
Now we are ready to submit the simulations to daint. Use, e.g.,
sbatch jobscript1.bash
to submit the first simulation in "pull1". Do the same in the folder "pull2" respectively.
You can use the command
squeue -u classXX
(substitute classXX by the username connected to your machine, look at the start of every line in the terminal) to check if your jobs are already running. If so, check the output-files in the folders ("pull1" and "pull2") for any errors. If your jobs are still queued, keep the terminal open and periodically check their status while working on part 1, so that errors can be caught early. If errors occur, don't hesitate to ask the tutor.
Start with part 1 of the tutorial, which you can entirely perform on the local machine (so use a new terminal).
Understanding the simulation setup
If necessary, log in to daint again. Check the status of your simulations again and have a look at the output-files. The simulations should be complete by now - preferably without errors.
If everything is fine, you need to copy the simulation results to your local machine for further analysis. On the cluster, first copy the folder containing the simulations to the home directory
cp -r /scratch/snx3000/classXX/gromacs_tutorial ~/.
Then open a new terminal on your local machine and copy the files
scp -r classXX@ela.cscs.ch:~/gromacs_tutorial ~/.
Finally, you should delete gromacs_tutorial from the home-directory of the cluster.
Should the simulations have failed for any reason, you can try to fix the problem and run again or, if there is a lack of time, download prepared simulation results of pull1 and pull2.
All the remaining steps are ment to be performed in your local copy of gromacs_tutorial. You can logout from the cluster.
Question: With your knowledge from part 1 of the tutorial, can you figure out what the files "start.gro", "topol.top", "index.ndx", as well as the .mdp-files are good for?
Question: Look at "pull1.mdp" and "pull2.mdp". At which parameters do the two simulations differ? The pulling-options are set at the bottom of each file. Use the gromacs manual to find out where the pulling forces were applied in the simulations, which spring constants and which pulling speeds were used.
Analyzing the simulation results
Inspect both simulated trajectories visually using VMD, e.g. with
vmd start.gro pull1/pull1.xtc
The selection-keywords "protein" and "resname BTN" will be useful; also change the drawing methods to something appropriate, e.g., "New Cartoon" for the protein and "Licorice" for biotin.
Question: Do you see the unbinding of biotin from streptavidin? Can you spot intermediate binding states in the trajectory with the slower pulling?
For both simulations, the pulling forces were recorded in "pull1/pull1_pullf.xvg" and "pull2/pull2_pullf.xvg", respectively. Plot both of them versus the simulated time, e.g., with
xmgrace pull1/pull1_pullf.xvg
and determine the rupture forces.
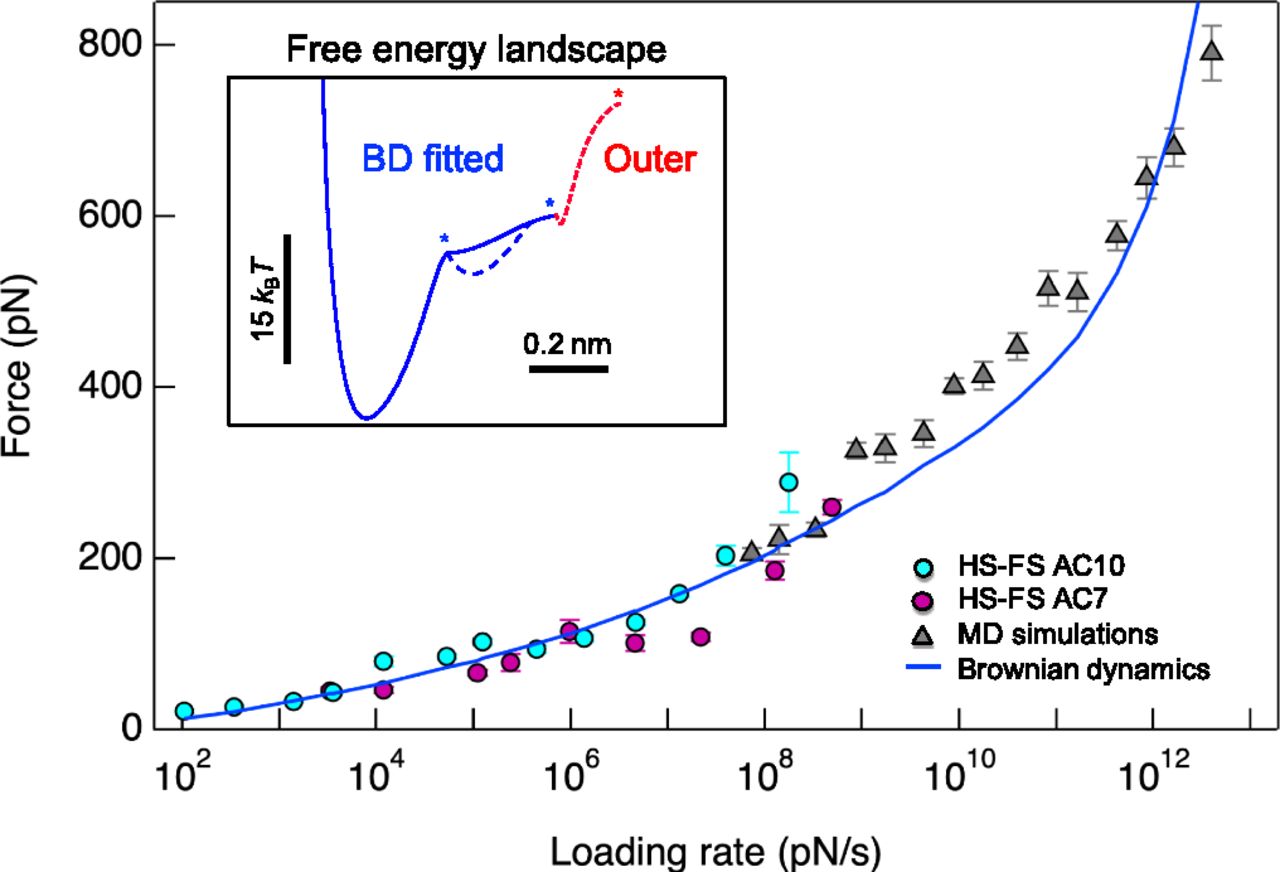
On the right you see a plot of the rupture forces depending on the loading rate from experiments, brownian dynamics and MD simulations from Rico et al. [Proc. Nat. Acad. Sci., 2019]. Calculate the loading rate (product of the spring constant and the pulling velocity) for each of your simulations and compare your results to the figure. Hint: Gromacs standard units are nm for distances, ps for times and kJ/mol for energies.
Question: How do your results compare to those in the figure? What could be possible reasons for deviations?
Question:
Why is it so difficult to achieve overlap between simulation results and experiments for pulling?
Further references and advanced reading